






JMLS 2021 December;6(2):97-105. 10.23005/ksmls.2021.6.2.97 Epub 2021 December 15
Copyright © 2021 by The Korean Society of Marine Life Science
Chromosomal Assembly of Tegillarca granosa Genome using Third-generation DNA Sequencing and Hi-C Technology
Jinmu Kim; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Seung Jae Lee; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Euna Jo; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea; Unit of Research for Practical Application, Korea Polar Research Institute (KOPRI), Incheon 21990, Korea
Eunkyung Choi; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Minjoo Cho; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
So Ryung Shin; Department of Aqualife Medicine, Chonnam National University, Yeosu 59626, Korea
Jung Sick Lee; Department of Aqualife Medicine, Chonnam National University, Yeosu 59626, Korea
Hyun Park; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
- Abstract
꼬막은 해양 어업으로써 아시아 전 지역에 있어서 중요한 수산자원 중 하나이다. 하지만, 공장의 산업화, 해양 환경오염, 그리고 지구 온난화로 인해 해양 어업 생산량이 급격히 떨어졌다. 우리나라 남해안의 주요 수산자원인 꼬막의 유전적 특성을 파악하기 위하여 꼬막의 전장유전체를 해독하고 염색체 서열을 규명하였다. 915.4 Mb의 게놈을 조립하였고, 19개의 염색체 유전자 서열을 식별하였다. 꼬막의 유전체에서 25,134개의 유전자들을 확인하였고, 그 중에 22,745개의 유전자들에 대한 기능을 확인했으며, 4,014개의 유전자들에 대한 KEGG pathway를 분석하였다. 꼬막유전체와 8종의 다른 패류와 비교유전체 분석을 통하여 확장/감소(gene gain and loss) 분석을 수행한 결과, 725개의 유전자군의 확장과 479개의 유전자군의 감소를 확인하였다. 꼬막의 homeobox 유전자 클러스터는 촉수담륜동물 내에서 잘 보존된 유전자 구조를 보였다. 또한, 꼬막은 3개의 hemoglobin 유전자들이 피조개의 hemoglobin과 높은 유사성을 보였다. 꼬막의 전장유전체 정보를 통해 꼬막의 환경 적응과 진화의 유전적 특성과 생리적 특성뿐만 아니라, 꼬막 양식의 효율성을 높이는 양식산업에 널리 이용될 수 있는 유전적 정보를 제공할 것이다.
Tegillarca granosa, is one of the most important fishery resources throughout Asia. However, due to industrialization factories, marine environmental pollution, and global warming, the marine fishery production has drop sharply. In order to understand the genetic factors of the blood clam, which is a major fishery resource on the southern coast of Korea, the whole genome of blood clam was studied. The assembled genome of T. granosa was 915.4 Mb, and 19 chromosomes were identified. 25,134 genes were identified, and 22,745 genes were functionally annotated. As a result of performing gene gain and loss analysis between the blood clam genome and eight other types of shellfish, it was confirmed that 725 gene groups were expanded, and 479 gene groups were contracted. The homeobox gene cluster of blood clam showed a well-preserved genetic structure within lophotrochozoan ancestor. T. granosa genome showed high similarity between three hemoglobin genes with Scarpharca broughtonii. The blood clam genome will provide information for the genetic and physiological characteristics of blood clam adaptation, evolution, and the development of aquaculture industry.
Keywords: Tegillarca granosa(꼬막) Gene gain-and-loss analysis(유전자 확장/감소 분석) Homeobox(호메오박스) Phylogenetic tree(계통수) Hemoglobin(헤모글로빈)
Correspondence to: Hyun Park; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
- Received
- 18 November 2021;
- Revised
- 18 November 2021;
- Accepted
- 25 November 2021.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Language: Korean/English,
Full Text:

서 론
꼬막(Tegillarca granosa)은 연체동물문에서 이매패강 중 돌조개과에 속하는 종으로서, 주로 인도 태평양 연안의 갯벌과 동아시아에 서식하고, 동남아시아의 갯벌에서 넓게 분포하고 있다. 또한, 한국, 중국, 일본의 서태평양 연안에서 수산업에 있어서 매우 중요한 자원이고 많은 생산량을 차지하고 있다(Liu et al., 2017). 하지만, 최근에 환경오염, 다양한 수산질병 발생 및 지구 온난화로 인하여 꼬막의 최대 생산지역인 아시아에서 조개 어업에 심각한 문제를 겪고 있다(Abbas Alkarkhi et al., 2008). 이러한 문제점들을 해결하면서 어업 생산량을 증가시키기 위해 T. granosa 및 굴, 홍합, 가리비와 같은 다양한 패류의 유전적 정보와 전사체의 정보에 대해서 연구가 다양하게 진행이 되고 있다(Bai et al., 2019).
수산양식의 생산을 더 효율적이고 안전하게 만들기 위해 다양한 유전적 연구가 수행되고 있다. 패류는 바이러스나 박테리아에 의해 손상되고 감염되면, 면역세포로 간주되고 있는 혈구세포가 면역방어 반응에 관여하게 된다(Bao et al., 2011). 패류의 혈구 또는 순환세포로 불리는 것은 리소좀 효소, 독성 산소 중간체 및 다양한 항균 펩타이드와(Canesi et al., 2002) 같은 병원균 및 기생충에 대한 방어 메커니즘을 담당하게 된다. 대부분 패류의 혈구에는 붉은색을 나타내는 hemoglobin이 없지만 피조개(Scapharca broughtonii)와 (Bai et al., 2019) 같은 종에 대해서 붉은색을 띄는 hemoglobin이 발견되었다. Hemoglobin은 프로피린 고리의 중심에 있는 철 원자와 폴리펩타이드 사슬의 히스티딘 잔기 사이에 산소 및 기타 기체 리간드를 결합하게 해주는 heme 단백질을 갖고 있으며(Abbas Alkarkhi et al., 2008), 이를 통해, 신체 내에 산소를 공급함으로써 유산소 신진대사를 도와준다(Jiang et al., 2007). 최근에 hemoglobin의 항균 활성이 Scapharca subcrenata 및 T. granosa에서 보고되었지만(Liu et al., 2017) 아직까지도 근본적인 원인에 대해서 여전히 확인이 되지 않았다(Wang et al., 2021).
다양한 패류의 유전적 정보와 관련하여 또 다른 관심분야로 연구되고 있는 부분은 hox 유전자이다. Hox 유전자는 신체의 올바른 위치에 올바른 구조가 형성되도록 위치의 특성을 암호화하며 지정하게 된다. 예를 들어, 곤충의 hox 유전자는 분절에 어떤 부속기관이 형성되는지 지정하게 되며 척추동물에서는 hox 유전자가 척추의 유형과 모양을 지정하게 된다(Barucca et al., 2003). 또한, hox 유전자는 전사 인자를 암호화하고 진화적으로 생물 내에 잘 보존되어 있다(Hajirnis and Mishra, 2021).
본 연구에서는 꼬막의 전장유전체(whole-genome sequencing)를 3세대 염기서열 기술을 이용하여 해독하고, 꼬막의 유전체 분석을 기반으로 피조개와 hemoglobin 유전자들에 대한 비교 및 패류 내에 hox 유전자들에 대한 유전적 구조를 비교분석 하였다. 본 연구에서 제시된 꼬막의 전장유전체 정보는 꼬막의 환경 적응, 진화의 유전적 특성 및 생리적 특성뿐만 아니라 꼬막 양식의 효율성을 높이는 양식산업에 널리 이용될 수 있는 유전적 정보를 제공할 것이다.
재료 및 방법

1. 시료 및 유전자 추출
T. granosa는 2019년 9월 대한민국 순천 벌교에서 채집했으며(Fig. 1A), 꼬막의 근육 조직에서 DNA를 phenol: chloroform: isoamyl alcohol 방법을 이용하여 추출하였다. DNA의 정량 및 품질 확인을 위해서 Qubit 2.0 fluorometer (Invitrogen, Life Tech- nologies, CA, USA)와 BioAnalyzer (Agilent Technologies, CA, USA)을 이용하여 확인하였다.
RNA 추출을 위하여 꼬막의 아가미, 혈액, 비장, 난소, 소화관을 해부하여 액체질소에 즉시 냉동하였고 -80℃에 보관하였다. RNA 추출은 TRIzol (Invitrogen, USA)을 이용하여 제조사의 프로토콜에 따라 추출하였다. RNA 정량은 Qubit 2.0 Fluorometer (Thermo- Fisher Scientific, USA)를 사용하여 확인하였고, RNA의 integrity에 대해서 Fragment Analyzer Systems (Agilent Technologies, USA)을 사용하여 확인하였다. RNA 샘플에 대한 표준 RNA 품질은 RIN > 7.0 (RNA Integrity Number)이였으며, 2.0 < O.D. 260/280 < 2.2 RNA 샘플에 대해서 PacBio와 Illumina 라이브러리를 제작하여 시퀀싱에 사용되었다.
2. PacBio Sequencing 및 Hi-C Sequencing
PacBio 플랫폼을 이용한 DNA 염기서열 분석을 위하여, SMRT bell (single-molecule real-time sequencing) 라이브러리를 구축하였고, 이를 만들기 위해, PacBio DNA Template Prep Kit 1.0 (Pacific Biosciences, Menlo Park, CA, USA)을 사용하였다. 각각의 라이브러리에 대해 2100 Bioanalyzer (Agilent Technologies)을 이용하여 품질과 정량을 확인하였다. 제작한 DNA 라이브러리는 SMRT cell (Pacific Biosciences, Sequel SMRT Cell 1M v2)을 사용하여 시퀀싱하였고, 총 10개의 SMRT cell을 사용하였다.
꼬막의 근육 조직을 이용하여 염색체 기반의 Hi-C chromatin contact map을 제작하기 위해 Illumina 라이브러리를 구축하였다. Hi-C 분석을 위한 근육 조직 고정, 염색질 분리 및 라이브러리 구성은 제조사의 방식에 의해 수행되었다(Belton et al., 2012). 생성된 라이브러리의 농도, 삽입 크기 및 적절한 농도를 확인한 후 최종 라이브러리를 사용해서 Illumina NovaSeq 플랫폼(Illumina, USA)에 시퀀싱하여 데이터를 생산하게 되었다.
3. RNA sequencing
각 기관별 추출한 RNA는 SMARTer PCR cDNA Synthesis Kit (Clontech, Japan)을 사용하여 cDNA로 합성하였다. 1~5 μg의 풀링된 cDNA을 사용하여 라이브러리를 제작 후, 총 2개의 SMRT cell을 PacBio Sequel system (Pacific Bioscience)을 사용하여 분석하였다. 시퀀싱된 데이터는 SMRT link (ver. 6.0)을 이용하여 필터링 및 클러스터링을 수행하여 full-length cDNA를 추출하였다.
4. 꼬막의 게놈 조립 및 유전자 주석
꼬막유전체의 de-novo assembly을 위해, FALCON-Unzip assem- bler (ver. 0.4)을 사용하였다(Chin et al., 2016). 게놈 조립 품질을 향상시키기 위해 FALCON-Unzip assembler는 정렬되지 않은 BAM 파일을 raw data로 사용하고, 기존에 디폴트 값으로 되어 있는 알고리즘을 사용하여 파일을 정제하였다.
FALCON-Unzip을 사용한 후, Hi-C 라이브러리를 사용하여 3D-denovo 조립(3D-DNA) 파이프라인에 있는 디폴트 파라미터를 이용해서 꼬막의 scaffold에 mapping하였다(Dudchenko et al., 2017). 꼬막의 유전체 크기는 K-mer 기반의 기존 연구를 이용하여 예측하였다(Kim et al., 2021).
꼬막의 염색체 수준 어셈블리의 완성도를 확인하기 위해, 어셈블된 scaffold를 Benchmarking Universal Single-Copy Orthologs (BUSCO) (ver. 3.0)을 사용하였다. BUSCO에서 사용된 리니지 데이터 세트 옵션은 metazoan odb9 데이터 베이스를 사용하였다(Manni et al., 2021).
다음으로, RepeatModeler (ver. 1.0 RRID: SCR_015027)에 있는 디폴트 파라미터를 사용하여, de novo repeat 라이브러리를 구축하였고, tandem repeats을 포함하여 simple repeats, satellites, 그리고 low-complexity repeats에 대해서 분류하였다(Bao and Eddy, 2002).
유전자 주석은 Maker (ver. 2.28)를 이용하였고, 총 3라운드의 반복 훈련을 사용하였다(Cantarel et al., 2008). 첫 번째 라운드에서 ab initio 유전자 예측은 Snap (Snap, RRID: SCR_002127) 및 Augustus (Augustus: Gene Prediction, RRID:SCR_008417)을 사용하였다. 또한, 전사체 정보로써 Iso-Seq 데이터와 NCBI에서 얻은 패류의 단백질 서열 정보를 사용하였다. 두 번째 라운드에서 유전자 예측을 위해 재 훈련된 HMMS를 사용하였다. 최종 라운드를 진행한 후, 그 결과는 꼬막의 유전자 예측에 사용이 되었으며, 본 데이터를 바탕으로 꼬막의 유전체 분석을 진행하였다.
Maker을 이용하여 나온 최종 데이터를 기반으로, 단백질 시퀀스는 NCBI nr database (E < 10-10)에 BlastP (ver. 2.2.29)를 사용하여 분석하였다. 단백질의 도메인 분석은 Interproscan5을 사용하였고, Blast2GO 기반의 유전자 온톨로지(GO) 분석(ver. 4.19) (Gotz et al., 2008)과 Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al. 2017) 데이터 베이스를 이용하여 생물학적 시스템의 기능 및 유틸리티를 확인하였다.
5. 염색체의 gene synteny와 유전자 확장/감소 분석
꼬막의 염색체와 피조개의 염색체 간에 gene synteny을 확인하기 위해 Mummer4 (Marcais et al., 2018)을 이용하여 염색체 서열을 정렬하였다. 꼬막과 피조개의 정렬된 염색체 서열을 사용하여 Circos (Krzywinski et al., 2009)을 통해 gene synteny를 비교하였다.
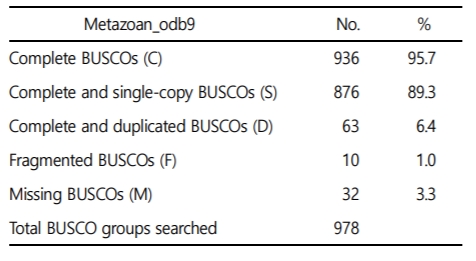
꼬막과 다른 패류 간 유전자 확장/감소(gene gain-and-loss) 분석을 위해 유전체 정보가 알려진 8종의 패류종의(Table 6) 정보를 이용하여 OrthoFinder (Emms and Kelly, 2019)을 사용하였다. OrthoFinder를 통해 나온 결과를 이용하여 9종의 패류 간 유전자 확장/감소를 확인하였고, 9종의 패류에 해당되는 single-copy orthologue 서열들을 이용하여 MegaX (Kumar et al., 2018)을 통해 계통도를 완성하였다. 꼬막을 포함한 9종 간의 divergence time을 확인하기 위해 TimeTree (Kumar et al., 2017)을 이용하여 Crassostrea gigas와 Pinctada fucata martensii 종의 divergence time 기점으로 다른 종에 대해서도 예측하였다(Fig. 1D).
6. Hox 및 hemoglobin 유전자 비교분석
9종의 패류 게놈 데이터 바탕으로(Table 6), 촉수담륜동물 조상에서 나타난 parahox와 hox 클러스터를 참고하여 꼬막 및 다른 패류 내에 hox 유전자들에 대한 gene synteny을 비교하였다. 각 클러스터에 해당되는 유전자들의 구조와 방향성을 확인하였으며, 꼬막과 피조개에서 hox 유전자를 구성하고 있는 염색체의 위치에 관해서도 확인하였다. 척추동물과 패류에서 hemoglobin 유전자를 가진 종에 해당되는 데이터를 사용하여 Mega X에서 bootstrap 500을 설정하고 계통도를 완성하였다. Hemoglobin 유전자를 가진 종과 myoglobin 유전자를 가진 종에 관한 NCBI accession number는 계통도에 나열하였다. 꼬막, 피조개, 그리고 척추동물들이 가지는 hemoglobin 유전자 사이에 진화적으로 잘 보존되어 있는지 확인하기 위해, hemoglobin 유전자들의 아미노산 서열을 ClustalW를 이용하여 비교분석하였다. 꼬막과 피조개 사이에 hemoglobin 유전자에 관한 gene synteny을 확인하기 위해 염색체 수준에서 비교분석 하였다. Hemoglobin 및 neuroglobin 유전자를 포함하여 다른 유전자들에 대한 방향성을 확인하였고, 유전자의 삽입 및 삭제 그리고 두 종 사이에 hemoglobin 유전자를 구성하고 있는 염색체의 위치에 관해서도 분석하였다.
결 과

1. 꼬막의 게놈 시퀀싱 및 염기서열 분석
꼬막의 게놈 조립을 수행하기 위해 PacBio Sequel 플랫폼을 이용해서 10개의 SMRT cell을 사용하여 101.4 Gb의 유전자 서열을 생산하였다. 또한, 꼬막의 염색체 scaffolding을 위하여 NovaSeq 플랫폼을 사용하여 총 23.09 Gb의 Hi-C 라이브러리를 생산하였다. 마지막으로, 전사체 정보 분석을 위하여 PacBio Sequel 플랫폼을 이용하여 Iso-Seq 데이터 13.1 Gb를 생산하였다(Table 1).
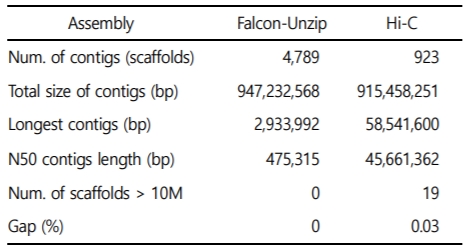
유전체 조립은 long-read 서열을 이용하여 FALCON-Unzip assembler를 이용하였다. 조립된 게놈의 총 크기는 947 Mb였고, N50 길이가 475,315 bp로 확인되었다. 다음으로, Hi-C super-scaffolding을 수행하여 최종적으로 915.4 Mb의 완성된 게놈 서열을 얻었으며, N50 길이가 총 45.6 Mb로 산출되었다(Table 2). 조립된 꼬막의 유전체로부터 총 19개의 염색체 서열이 확인되었다. 염색체 길이의 합은 823.2 Mb이며, 90 Mb의 서열은 염색체에 포함되지 않았다(Table 3, Fig. 1B).
조립된 유전체의 완성도를 평가하기 위해, BUSCO 분석은 metazoan odb9 데이터 세트를 통하여 확인하였다. 전체적으로 978개의 BUSCO 그룹이 확인되었으며, complete BUSCO는 95.7%로 확인되었다(Table 4).
2. 꼬막의 유전자 주석 분석
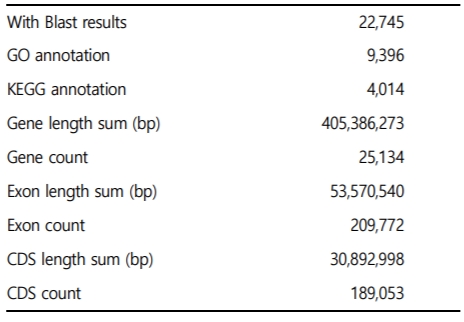
꼬막의 유전자 주석을 위하여 조립한 유전체 서열은 Repeat- Modeler를 사용해서 게놈 안에 있는 반복요소를 식별하고, 주석을 위한 반복 라이브러리를 생산하였다. 꼬막의 유전자 주석 분석은 꼬막전사체 Iso-Seq 데이터와 근연종의 단백질 서열을 사용하여 Maker를 이용하여 총 3번의 라운드를 통해, 총 25,134개의 유전자에 대한 structural annotation이 분석되었다. 유전자 기능분석은 NCBI nr database에 BlastX를 통하여 유전자를 정의하고, Blast2GO 프로그램을 통하여 유전자 온톨로지(GO) 분석 및 KEGG 분석을 수행한 결과, 22,745개의 유전자가 확인되었고, 그 중 9,396개는 GO mapping 되었으며, 4,014개의 유전자는 KEGG pathway에 mapping 되었다(Table 5).
3. Gene synteny와 gene gain-and-loss
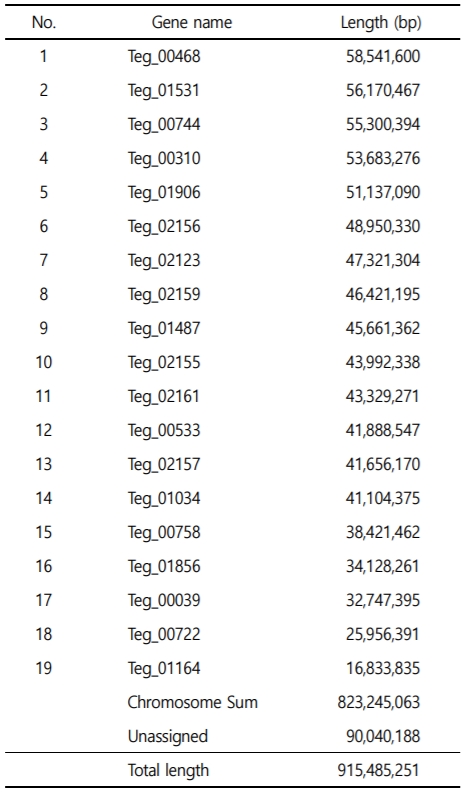
꼬막과 피조개의 염색체에서 유전자 구조적 상동성을 확인하기 위해 Mummer4를 이용하여 염색체 서열을 정렬하였다. 정렬된 염색체 서열은 Circos를 이용하여 시각화 하였고, 두 종 간 염색체 사이에서 높은 상동성이 확인되었다(Fig. 1C).
꼬막 및 다른 패류 사이에 유전자 확장/감소 분석을 위해 게놈 조립이 완료된 9종에 대한 단백질 서열 바탕으로(Table 6) OrthoFinder를 이용하여 상동유전자를 분석하였다. 9종의 패류에서 single copy orthologs는 1,351개로 확인되었으며, 꼬막에서 multiple copy orthologs, un-clustered gene, unique paralogs는 4,471개, 2,895개, 그리고 1,479개 순으로 확인되었다(Fig. 1D). 1,351개의 single copy orthologs 서열을 이용하여 9종에 해당되는 패류의 계통도를 Mega X를 통해 완성하였고, C. gigas와 P. fucata martensii 종의 divergence time을 기점으로 꼬막 및 다른 종의 divergence time을 예측하였다. 꼬막과 피조개는 90.21 Mya (million years ago)의 분기시점을 가진 것으로 확인되었다(Fig. 1D).
패류 9종에 대한 유전자 확장/감소 분석결과, 꼬막에서 725개의 유전자가 확장되었고, 479개의 유전자가 감소되었다. 9종 중에 Lingula anatina가 1,400개의 유전자로 가장 높게 확장되었으며, Pecten maximus가 151개로 가장 낮게 확장되었다(Fig. 1D).
4. Hox 및 hemoglobin 유전자 비교분석
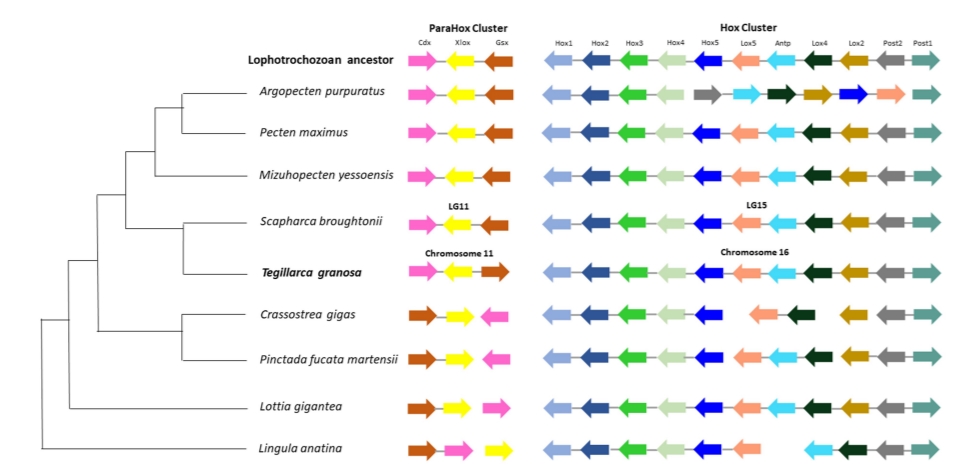
촉수담륜동물 조상에 있는 parahox와 hox 클러스터를 참고하여 9종에 해당되는 패류의 hox 유전자에 대한 유전적 구조를 비교분석 하였다. 9종에 해당되는 계통도는 Fig. 1D를 참고하였고, 피조개에서 parahox 클러스터는 LG11에 존재했으며, 꼬막에서는 11번 염색체에서 확인되었다. Hox 클러스터에 관여하는 유전자들에 대해서는, 꼬막은 16번 염색체에서 확인되었으며, 피조개에서는 LG15에 존재하였다. 촉수담륜동물 조상의 parahox와 hox 클러스터 바탕으로 9종의 패류에서 homeobox 유전자 클러스터가 잘 보존되어 있는 것으로 확인되었다(Fig. 2).
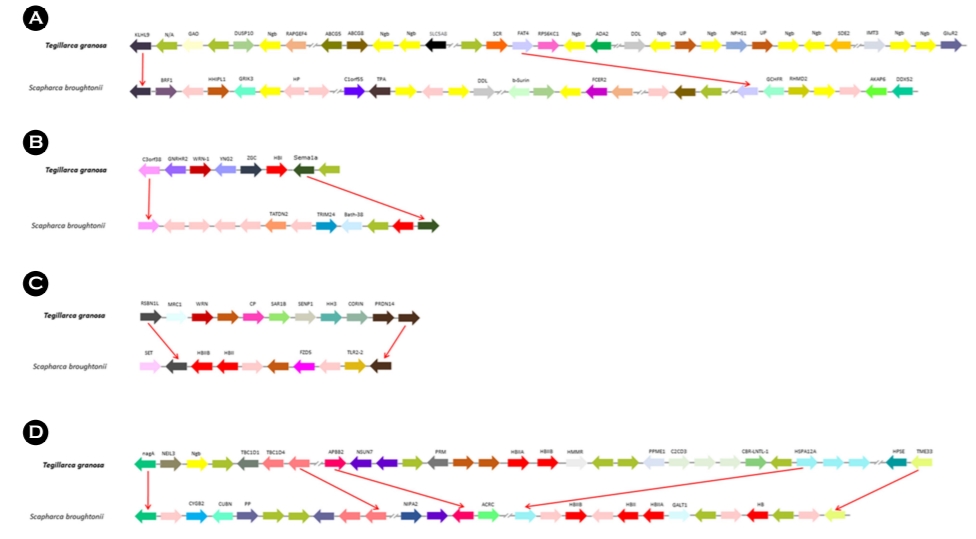
패류 및 척추동물에서 hemoglobin 유전자를 가진 단백질 서열을 이용하여 Mega X를 통해 계통도를 확인하였으며, 꼬막에서 hemoglobin 관련 유전자들은 피조개가 갖고 있는 hemoglobin 유전자와 같은 범주로 묶이는 것으로 확인되었다(Fig. 3A). 또한, 꼬막, 피조개 및 척추동물의 hemoglobin 아미노산 서열을 ClustalW를 이용하여 비교분석 한 결과, 30~49 bp, 64~81 bp, 90~113 bp, 그리고 167~179 bp 사이에 있는 아미노산 서열들이 척추동물 및 꼬막과 피조개에서 잘 보존되어 있는 것으로 확인되었다(Fig. 3B). Hemoglobin 유전자를 가진 피조개와 꼬막 사이에 gene synteny를 확인하였다. Fig. 4A에서 꼬막과 피조개의 neuroglobin는 각각 11번 염색체와 LG11에서 확인되었고, 꼬막에서 11개의 neuro- globin이 존재했으며, 피조개에서는 5개의 neuroglobin이 관찰되었다. Fig. 4B에서 HBl는 꼬막에서 7번 염색체에서 관찰이 되었고, 피조개에서는 LG5에서 확인되었으며, 2종에서 각각 1개씩 식별되었다. Fig. 4C에서 HBllB와 HBll는 피조개의 LG6에서만 확인되었고, 꼬막에서는 식별이 되지 않았다. Fig. 4D에서는 다른 hemoglobin 유전자들이 꼬막의 11번 염색체에 존재했으며, 피조개에서는 LG11에 존재하는 것으로 확인되었다.
고 찰
꼬막은 아시아 전역의 해양 어업을 위한 보편적이고 주요한 자원이다. 이 종은 주로 얕은 해안과 갯벌에 서식하며, 다른 패류와 다르게 혈액에 hemoglobin을 갖고 있으며, 이와 관련된 다양한 연구들이 활발히 진행 중이다(Liu et al., 2017). 본 연구에서는 꼬막의 19개의 염색체를 포함한 전체 게놈 정보를 해독하였다. 해독된 꼬막의 유전체를 기반으로 8종의 패류들의 유전체 정보와 비교분석 한 결과, 이들 사이의 총 1,351개의 single copy orthologs가 확인되었으며, 꼬막은 종 특이적으로 725개의 유전자가 확장되었고, 479개의 유전자가 감소된 것을 확인하였다.
Hox 유전자는 신체의 올바른 위치에 대한 구조를 형성하도록 위치의 특성을 암호화하여 저장하게 되고, 종 간에 진화적으로 잘 보존되어 있다(Hajirnis and Mishra, 2021). 촉수담륜동물 조상의 parahox와 hox 클러스터의 유전자들 바탕으로 9종의 패류에 해당되는 homeobox 유전자 클러스터를 완성하였고, 그 중에 꼬막의 parahox 클러스터는 11번 염색체에 존재하는 것으로 확인되었다. Hox 클러스터는 16번 염색체에 존재했으며, 꼬막과 피조개 사이에 parahox 클러스터에는 Gsx 유전자에 대한 방향성이 서로 달랐으며, hox 클러스터를 구성하는 11개의 유전자에 대한 방향성이 모두 동일하였다. 이를 통해 두 종 간 homeobox 유전자 클러스터는 잘 보존되어 있는 것으로 확인되었다.
다양한 패류의 연구분야 중 하나인, 혈구는 순화세포로 불리고, 이를 이용하여 다양한 항균 펩타이드, 독성 산소 중간체 및 리소좀 효소와(Canesi et al., 2002) 같은 병원균 및 기생충에 대한 방어 메커니즘을 담당하게 된다. 반면, 꼬막의 혈구에는 대부분 hemoglobin으로 구성되어 있고, 이는 붉은색을 띄게 해주는데(Abbas Alkarkhi et al., 2008), 이를 이용하여 신체 내에 산소를 공급함으로써 유산소 신진대사를 도와주게 된다(Jiang et al., 2007). 최근 연구에서는 항균 활성에 관한 부분이 보고되었지만, 아직까지 근본적인이 원인에 대해서는 확인되지 않았다(Wang et al., 2021). 꼬막의 게놈 조립을 통해, hemoglobin 유전자를 가지고 있는 패류 및 척추동물의 서열을 이용하여 계통도를 완성하였고, 꼬막에 존재하는 hemoglobin 관련 유전자들은 Scapharca kago- shimensis, S. broughtonii와 Barbatia lima 종과 유사한 것으로 확인되었다(Fig. 3A). 피조개의 hemoglobin 유전자와 gene synteny을 비교분석 한 결과, neuroglobin 유전자는 꼬막에서 11번 염색체에서 11개가 확인되었으며, 피조개에서는 총 5개의 neuroglobin이 LG11에서 확인되었다. 꼬막과 피조개의 염색체 내에 neuroglobin 외에 다양한 hemoglobin 유전자와 관련하여 gene synteny를 완성하였고(Fig. 4), 유전자들 사이에 삽입과 삭제가 이루어지면서 정확한 원인에 대해서는 확인이 되지 않지만, 두 종 사이에 유전적 변이를 통해 생물학적 다양성이 존재할 것이라고 예상하였다(Barton and Zeng, 2019).
대부분의 꼬막 생산량을 이루고 있는 순천 및 여수 갯벌은 최근 폐수, 중금속 과다노출, 그리고 온도변화로 인하여 심각한 환경적인 문제를 겪고 있다. 이와 관련하여, 본 연구에서 해독한 꼬막의 유전체 정보 기반으로, 중금속 노출 및 온도변화에 따른 다양한 유전자 발현변화를 확인하여, 향후 꼬막의 양식 효율성을 높이기 위한 기초자료를 제공할 것이고, 이 외에도 꼬막의 생리, 생화학적 연구, 환경적응성과 진화 특성에 대한 연구 기초자료를 제공할 것이다.
- References
-
1. Abbas Alkarkhi FM, Ismail N, Easa AM. 2008. "Assessment of arsenic and heavy metal contents in cockles (Anadara granosa) using multivariate statistical techniques". Journal of Hazardous Materials 150: 783-789.
-
2. Bai CM, Xin LS, Rosani U, Wu B, Wang QC, Duan XK, Liu ZH, Wang CM. 2019. "Chromosomal-level assembly of the blood clam, Scapharca (Anadara) broughtonii, using long sequence reads and Hi-C". Gigascience 8.
-
3. Bao Y, Wang Q, Lin Z. 2011. "Hemoglobin of the bloody clam Tegillarca granosa (Tg-HbI) is involved in the immune response against bacterial infection". Fish & Shellfish Immunology 31: 517-523.
-
4. Bao Z, Eddy SR. 2002. "Automated de novo identification of repeat sequence families in sequenced genomes". Genome Res 12: 1269-1276.
-
5. Barton HJ, Zeng K. 2019. "The Impact of Natural Selection on Short Insertion and Deletion Variation in the Great Tit Genome". Genome Biol Evol 11: 1514-1524.
-
6. Barucca M, Olmo E, Canapa A. 2003. "Hox and paraHox genes in bivalve molluscs". Gene 317: 97-102.
-
7. Belton JM, McCord RP, Gibcus JH, Naumova N, Zhan Y, Dekker J. 2012. "Hi-C: a comprehensive technique to capture the con- formation of genomes". Methods 58: 268-276.
-
8. Canesi L, Gallo G, Gavioli M, Pruzzo C. 2002. "Bacteria-hemocyte interactions and phagocytosis in marine bivalves". Microsc Res Tech 57: 469-476.
-
9. Cantarel BL, Korf I, Robb SM, Parra G, Ross E, Moore B, Holt C, Sanchez Alvarado A, Yandell M. 2008. "MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes". Genome Res 18: 188-196.
-
10. Chin CS, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A, Dunn C, O'Malley R, Figueroa-Balderas R, Morales-Cruz A, Cramer GR, Delledonne M, Luo C, Ecker JR, Cantu D, Rank DR, Schatz MC. 2016. "Phased diploid genome assembly with single-molecule real-time sequencing". Nat Methods 13: 1050-1054.
-
11. Dudchenko O, Batra SS, Omer AD, Nyquist SK, Hoeger M, Durand NC, Shamim MS, Machol I, Lander ES, Aiden AP, Aiden EL. 2017. "De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds". Science 356: 92-95.
-
12. Emms DM, Kelly S. 2019. "OrthoFinder: phylogenetic orthology inference for comparative genomics". Genome Biol 20: 238.
-
13. Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A. 2008. "High-throughput functional annotation and data mining with the Blast2GO suite". Nucleic Acids Res 36: 3420-3435.
-
14. Hajirnis N, Mishra RK. 2021. "Homeotic Genes: Clustering, Modu- larity, and Diversity". Front Cell Dev Biol 9: 718308.
-
15. Jiang N, Tan NS, Ho B, Ding JL. 2007. "Respiratory protein-generated reactive oxygen species as an antimicrobial strategy". Nat Immunol 8: 1114-1122.
-
16. Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. 2017. "KEGG: new perspectives on genomes, pathways, diseases and drugs". Nucleic Acids Res 45: 353-361.
-
17. Kim J, Lee SJ, Jo E, Choi E, Kim HJ, Lee JS, Park H. 2021. "Genome Survey and Microsatellite Marker Selection of Tegillarca granosa". Journal of Marine Life Science 6: 38-46.
-
18. Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. "Circos: an information aesthetic for comparative genomics". Genome Res 19: 1639-1645.
-
19. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. "MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms". Mol Biol Evol 35: 1547-1549.
-
20. Kumar S, Stecher G, Suleski M, Hedges SB. 2017. "TimeTree: A Resource for Timelines, Timetrees, and Divergence Times". Mol Biol Evol 34: 1812-1819.
-
21. Liu B, Teng S, Shao Y, Chai X, Xiao G, Fang J, Zhang J, Wang C. 2017. "A Genetic Linkage Map of Blood Clam (Tegillarca granosa) Based on Simple Sequence Repeat and Amplified Fragment Length Polymorphism Markers". Journal of Shellfish Research 36: 31-40.
-
22. Manni M, Berkeley MR, Seppey M, Simao FA, Zdobnov EM. 2021. "BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes". Mol Biol Evol 38: 4647-4654.
-
23. Marcais G, Delcher AL, Phillippy AM, Coston R, Salzberg SL, Zimin A. 2018. "MUMmer4: A fast and versatile genome alignment system". PLoS Comput Biol 14: e1005944.
-
24. Wang S, Yu X, Zhang S, Jin H, Chen Z, Lin Z, Bao Y. 2021. "Cu2+ Inhibits the Peroxidase and Antibacterial Activity of Homo- dimer Hemoglobin From Blood Clam Tegillarca granosa by Destroying Its Heme Pocket Structure". Frontiers in Marine Science 8: 2296-7745.
Copyright (C) 2024. The Korean Society of Marine Life Science